Protein Structure Prediction Kit 1.4 使用說明手冊
http://par.cse.nsysu.edu.tw/~pspk/
前言
Protein Structure Prediction Kit
1.4(PSPK 1.4)是由國立中山大學資訊工程所平行處理實驗室團隊,歷經四年所開發,在蛋白質結構預測的領域,方法不斷推陳出新,本實驗室團隊亦接連改良PSPK的核心演算法,並設計友善的使用者介面,以提供從事蛋白質研究的人一個好用的輔助工具為己志,本軟體免費提供非商業性學術研究使用,若有軟體操作及核心演算法等相關問題,歡迎各界不吝指教。
Department of Computer Science and
Engineering
National Sun Yat-sen University Kaohsiung,
Taiwan 80424
URL: http://par.cse.nsysu.edu.tw
TEL: +886-7-5252000 ext. 4345
概觀
PSPK的設計目的是輔助我們針對一條胺基酸序列,利用結構為已知的蛋白質序列資料庫(如已存在蛋白質資料庫PDB的蛋白質結構),並且依結構排列(Structure Alignment)、建立晶格模型結構(Folding)、曲線比對(Curve Matching)與整合結果(Merging Result)四大步驟來預測此目標序列之蛋白質結構。在預測完成後,提供三維結構檢視與RMSD比對等方便研究進行之功能。本操作技術手冊內容包括PSPK1.4的安裝、程式流程及操作方法。
安裝
PSPK支援下列作業系統:
– Microsoft Windows 98
(Second Edition)
– Windows Me
– Windows NT 4.0 (Service
Pack 6)
– Windows 2000 (Service Pack
3)
– Windows XP Professional
(Service Pack 1)
安裝主程式:
將檔案解壓縮後,即可執行PSPK.exe主程式。
主程式下載網址:http://par.cse.nsysu.edu.tw/~pspk/PSPK.zip
安裝BLASTP:
Basic Local Alignment Search Tool (BLAST),是由National Center Biotechnology Information(NCBI http://www.ncbi.nlm.nih.gov/ )開發的免費工具,用來尋找區域相似序列,而blastp是其中一種做protein-protein間比對的工具,效能較使用dynamic programming的方法佳。當第一次執行PSPK時,必須安裝blastp,可以選擇手動安裝或經由PSPK自動安裝。
手動安裝步驟:
1.
下載blastp:由NCBI的網站或FTP 站下載。
http://www.ncbi.nlm.nih.gov/BLAST/download.shtm
ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/
2.
下載蛋白質資料:由NCBI的FTP 站下載pdbaa.gz
ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/
將blastp執行檔及pdbaa.gz解壓縮在blast目錄下。
3.
在命令提示字元執行:
formatdb -i pdbaa -p T -o T
其中
-i Input file(s) for
formatting (this parameter must be set)
[File In]
-p Type of file
T - protein
F - nucleotide [T/F]
Optional
-o Parse options
T - True: Parse SeqId and create indexes.
F - False: Do not parse SeqId. Do not create indexes.
自動安裝步驟:
Setting→Set Blastp→Create
圖一:自動安裝
Blastp更新:
手動更新步驟:
1.
下載蛋白質資料:由NCBI的FTP 站下載pdbaa.gz
ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/
pdbaa.gz解壓縮在blast目錄下。
2.
在命令提示字元執行:
formatdb -i pdbaa -p T -o T
自動更新步驟:
Setting→Set Blastp→Updata
圖二:自動更新
安裝本機資料庫(Local Database):
PSPK使用的蛋白質結構資料來自Protein Data Bank(PDB http://www.rcsb.org/pdb/index.html
),若要手動建立本機資料庫須下載其中的PDB格式的蛋白質資料:pdb[PDB name].ent.Z,經解壓縮、轉檔才能為PSPK使用。PSPK提供建立本機資料庫的功能,步驟如下:System→ Create Local Database進入圖三畫面,預設由PDB下載,連線後可選擇mirror整個PDB資料庫或僅下載特定年份資料,唯mirror整個PDB資料庫約須要8GB的磁碟空間,費時數小時方能完成。
圖三:安裝本機資料庫
目錄、檔案說明如下:
目錄/檔案
敘述
Blast
包括所有Blastp所需檔案
l
Data
-blosum45、blosum62、blosum80:Blastp使用之Score Matrix。
l
blastall.exe:Blastp主程式。
l
formatdb.exe:建立Blastp使用的資料庫。
l
formatdb.log、pdbaa.phr、pdbaa.pin、pdbaa.pnd、pdbaa.pni、pdbaa.psd、pdbaa.psi、pdbaa.psq:Blastp
目錄/檔案
敘述
資料庫元件。
l
pdbaa:解壓縮後的Blastp資料庫原始檔。
Data
預設的本機資料庫資料存放路徑
Demo
預設的檔案輸入路徑
ScoreMatrix 包含預設與自訂的Score Matrix
Setting
包含PSPK的設定資料
l
color.txt:使用者設定的顏色。
l
default_color.txt:預設顏色。
l
default_psps.conf:預設變數值設定。
l
psps.conf:使用者自訂變數值設定。
Temp
暫時存放從PDB下載之蛋白質結構資料
l
gzip.exe:解壓縮軟體,將下載之蛋白質結構資料解壓縮。
blastp.BAT
執行Blastp
glut32.dll BCB所需元件
gzip.BAT
執行gzip
PSPK.exe
主程式
程式流程圖:
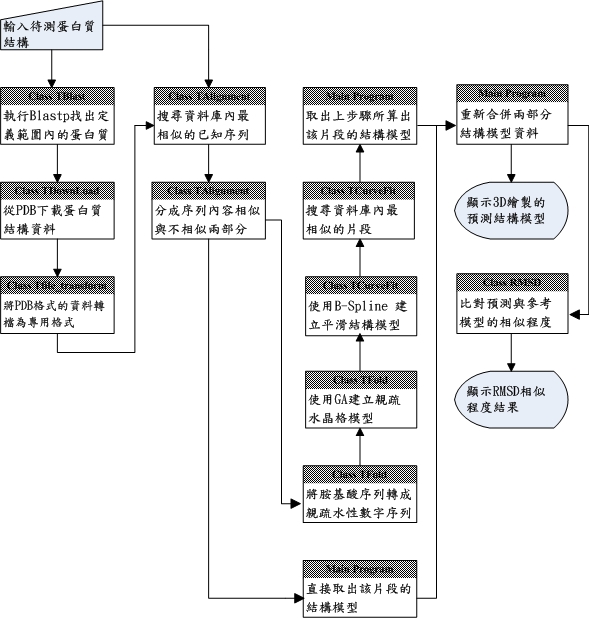
圖四:程式流程圖
操作方法
PSPK的核心演算法包括:「Prediction with Secondary Structure」,「Prediction without Secondary
Structure」,「Prediction on Sliced Lattice Model」三種,由使用者視需要選用。
STEP 1. Prepare:
選擇核心演算法Prediction with Secondary
Structure或Prediction
without Secondary Structure:
1.
資料輸入:若是Prediction with Secondary
Structure須輸入一級結構和二級結構序列,Prediction without Secondary
Structure則只要輸入一級結構序列,輸入模式可選擇用檔案或直接輸入蛋白質序列。
2.
資料庫:可選擇使用本機資料庫或網路連結PDB資料庫,若使用PDB資料庫,則需設定Identity範圍,PSPK將會下載與待測序列相似度在範圍內的蛋白質結構資料作為Database內的資料,使用者可藉由改變Identity範圍來控制Database內的資料量。
3.
Score
Matrix:除了選擇程式提供的Score Matrix,使用者尚可自訂所需的Score Matrix。
4.
摺疊(Fold)變數:使用者可自行調整基因演算法的世代數(Generation)、交配率(Crossover Rate)及突變率(Mutation Rate),以獲得最佳解。
5.
曲線吻合(Curve Fitting):使用者可自行決定建立B-spline曲線所欲插入點的數量。
6.
按下「Next Step」按鍵。
選擇核心演算法Prediction on Sliced Lattice
Model:
此種方法可選擇採用基因演算法或ACO演算法,若採用基因演算法,操作與Prediction without Secondary Structure方法相同,而採用ACO演算法則會出現一個設定畫面,其中變數皆可配合使用者需要做調整,以達結果最佳化。
STEP 2. Predict:
由設定畫面轉換成執行畫面,可藉由progress bar觀察程式運作進度,當程式執行完畢畫面會出現Alignment、Folding、Curve Fitting、Merging等四個部份的執行結果及其所佔時間比例。
STEP 3. Report:
Structure:
蛋白質3D結構顯示是由OPENGL(3D繪圖與模型程式庫)所繪製,可依據使用者喜好,訂定下列選項,顯示出欲求得之資訊,圖五為初始畫面:
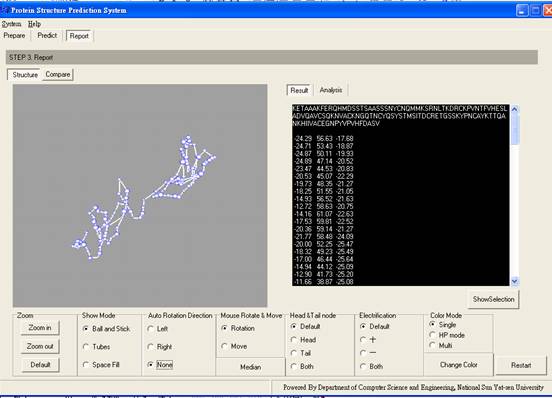
圖五:Report畫面
1.
Zoom:包含「Zoom in」、「Zoom out」及「Default」三個按鍵,供使用者將結構3D模型放大、縮小或是設成預設大小。
2.
Show
Mode:「Ball-Stick」、「Tubes」、「Space Fill」3種蛋白質結構表示方式,依使用者喜好自行選定。
3.
Auto
Rotation Direction:供使用者選定3D模型自動旋轉方向或旋轉與否。
4.
Mouse
Move & Mouse Rotate:讓使用者在畫布上用滑鼠拖曳3D模型,使其可以任意旋轉角度或是調整位置。
5.
Head
& Tail node:當結構趨於複雜時,點選此項可用顏色標示蛋白質結構的開頭和結尾胺基酸,分別為紅點和綠點如圖九。
6.
Electrification:以顏色標示胺基酸帶電性,「+」帶正電,以紫色示之,「-」帶負電,以黃色示之,電中性以藍色示之。如圖六所示。

圖六:標示胺基酸帶電性
7.
Color
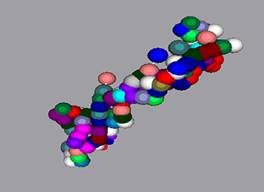
Mode:有3種模式可供使用者選擇,「Single」所有胺基酸採單一顏色,「HP mode」疏水性(hydrophobic)以藍點示之,親水性(hydrophile)以紅點示之(圖七),「Multi」每種安基酸皆給予不同顏色(圖八),按下「Change Color」按鍵進入色票畫面,供使用者改變顏色(圖九)。
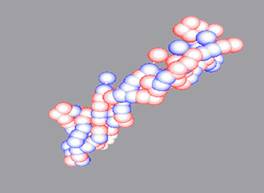
圖七:HP mode
圖八:Multi
圖九:Change Color
8.
Result:顯示胺基酸序列及各個胺基酸的座標位置,在胺基酸序列上按滑鼠左鍵反白選取,會在左邊的3D圖像上顯示出相對位置(圖十),此時若按下「Show Selection」按鍵,則左邊的3D圖像僅顯示使用者反白選取的那一段胺基酸序列,再按一次又會顯示全部(圖十一)。
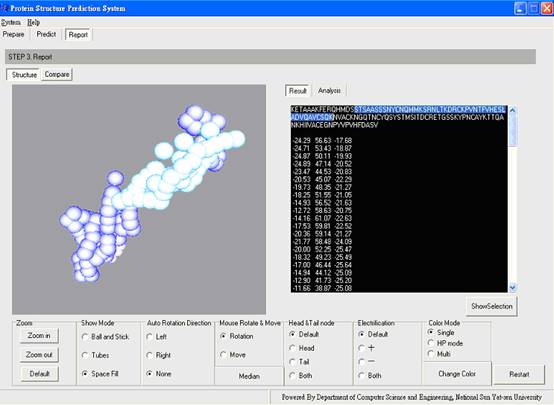
圖十:顯示選取序列在3D結構上的位置
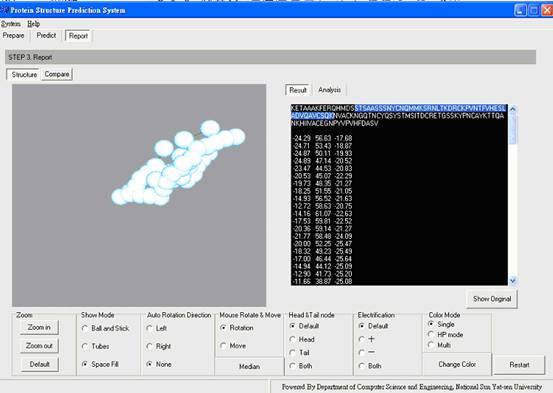
圖十一:只顯示選取序列
Protein
3D Viewer :
提供使用者可讀取RCSB提供的已知蛋白質結構檔案(副檔名為.ent or .pdb1)以3D模型顯示(圖十二)。
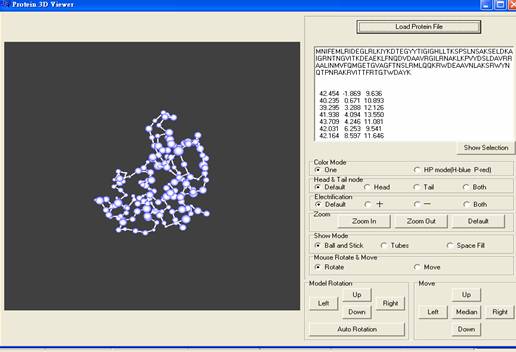
圖十二:Protein 3D Viewer
Compare:
用RMSD比對預測模型與參考模型的相似程度,先按「Load」鍵以檔案選取的方式讀進參考模型的結構資料,然後按「Calculate」按鍵計算RMSD值(圖十三)。
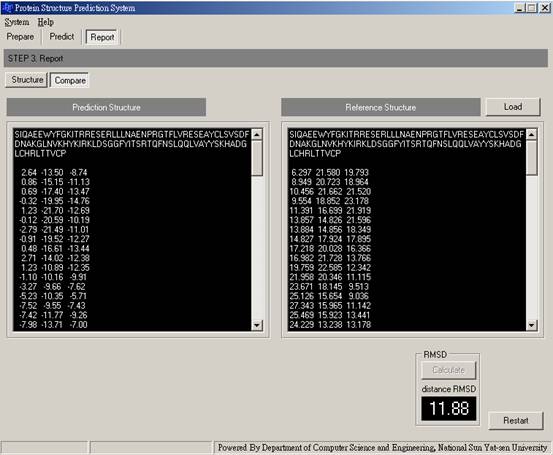
圖十三:用RMSD比對預測模型與參考模型的相似程度